The computations were made using an original Metropolis Monte Carlo code, which included a random number generator from Numerical Recipes, and built in statistical analysis which automatically detected when the system came into equilibrium and calculated the statistics from that point. We used a separate, similar code to calculate the free energy differences at the reference temperatures. The lattices were initialized with the molecules pointing to any point in the solid angle with equal probability, and all Monte Carlo steps were designed to ensure no preference of one orientation over another.
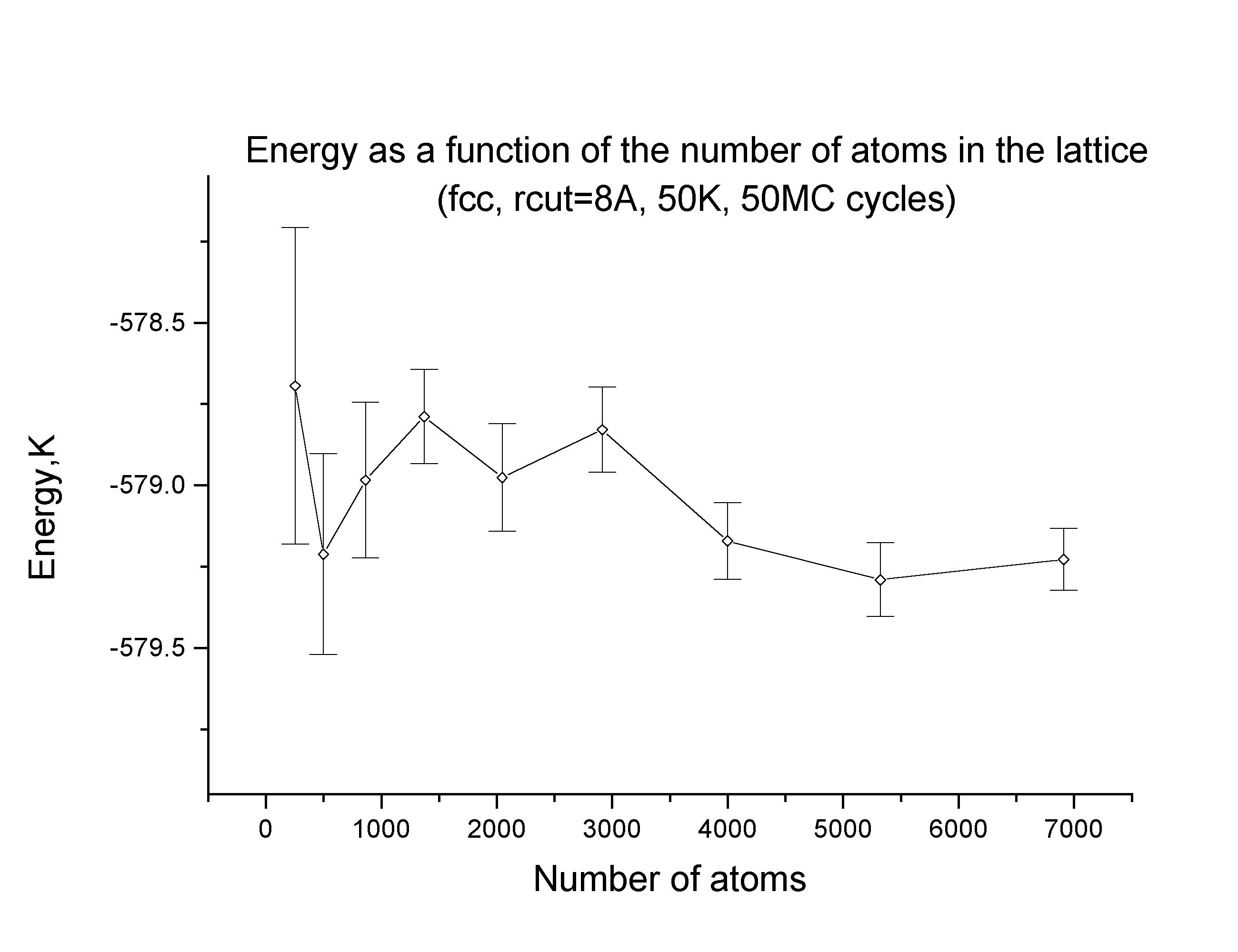
The simulation cell was taken to include 4000 atoms, large enough to reasonably eliminate unphysical correlation effects. The following plot shows the energy's dependence on the number of atoms in the simulation cell.
For neighbor tables we chose to include atoms within a cutoff radius of 8 angstroms (compared to nearest neighbor distances of approximately 4 angstroms). The dependence of the energies calculated on the cut-off radius is illustrated below.

With these approximations, the code took approximately 1 hour to run 200 Metropolis iterations per molecule on a DEC Alpha 500. While we would like to have taken a larger cut-off radius,we felt we could not afford the increase in required computer time. The resulting angular distribution at 30 K for the FCC phase shows that our simulation gave the proper orientational results for the FCC lattice, as seen below.

Next: Results
Up to Table of Contents